supertrees using tree ranking
The overall goal of the project is to summarize what is known about phylogenetic relationships in a transparent manner with a clear connection to analyses and the published studies that different clads. Comprehensive coverage of published phylogenetic statements is a very long term goal which would require work from a large community of biologists. The short-term goal for the supertree presented on the tree browser is to summarize a small set of well-curated inputs in a clear manner.
We hope that this tree (and its many deficiencies) will inspire biologists to help fix the tree by alerting us to relevant phylogenetic statements that are missing in our set of inputs or contributing to the Open Tree effort in other ways. We have built tools to support 2 types of feedback:
- Our phylogenetic study curator tool for adding new input trees.
- Clade-linked issue reports. Click on "See comments" on a node of the tree, such as the Tetrapoda node to see a conversation that is tied to a particular phylogenetic grouping. Full issue-tracking features for that conversation (e.g. notifications, authenticated comments, tagging, closing of the issue) are supported using GitHub issues system. This issue on GitHub contains the thread about the groupings shown at the Tetrapoda node linked above.
Of course, emailing comments is another way to provide feedback (see the opentreeofliife group for general comments and this list for software issues).
Note: MTH put together this list. #3 may not be agreed upon by all participants.
- produce a full supertree: The set of taxa associated with leaves of the supertree is the set of "terminals" in the taxonomic input.
- the tree should display (in the sense described here) as many edges from the curated input trees as possible . If the scoring criterion was a count of the number of groupings, then this would surely by an NP-hard problem REF needed. 1. ranked trees property: For simplicity of implementation, we use a tree-based ranking system to resolve conflicts for simplicity. 2. taxonomy last property: the taxonomy is the lowest ranked input
- Display no unsupported groupings sensu the definition describe in this doc.
- this one definitely needs some work: we might want some statement about the placement of groups that appear in only one source tree. Perhaps: when there is a large set of attachment points to which a group can be attached and still display the same set of input tree edges, then the group should be attached at the point closest to the root that is consistent with these displayed set. This placement is in the spirit of the Adams consensus of all of the attachment points.
- the tips of all input trees have been mapped to a common taxonomy (a pruned version of OTT in our case),
- if more than one species maps to the same OTT ID, then the tree has been pruned until this is not the case.
- if a tip maps to a higher taxon in OTT and another tip in the tree maps to taxon within the higher taxon, then one of the tips has been pruned (process repeated until the condition no longer applies).
- labels of internal nodes in the input phylogeny are ignored by the supertree algorithm.
- any redundant nodes (nodes of out-degree=1) have been suppressed.
- all trees are rooted.
The taxonomic input is a pruned version of OTT created by reading in OTT (2.8) and pruning taxa that have certain flags Needs documentation!
The strong preference for grouping based on tree-ranking simplifies the supertree construction problem considerably.
One very important processing step that treemachine does is remapping a tip in a tree to the deepest taxon that it could be considered to exemplify. MTH does not know if this step is discussed in any publication.
Operationally, this step consists of finding the deepest node in the taxonomy that is an ancestor of the tip's taxonomic label, but which is not an ancestor of any taxa that are the "outgroup" of that leaf with respect to the input tree.
To understand why this is important, consider the following inputs.
The highest ranked tree: 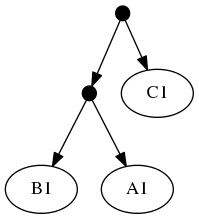
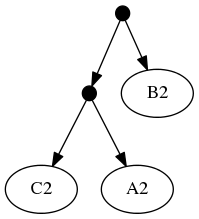
And the taxonomic tree:
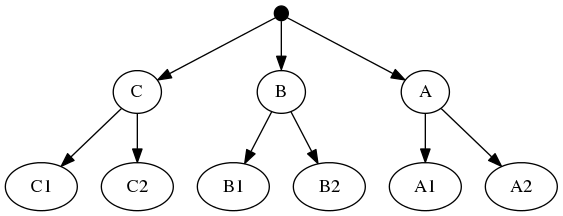
Note that the phylogenetic inputs have no overlap in their leaf sets. Thus, directly applyting the tree ranking score criterion (called WSITED on this page) to these inputs would return the following "phylo-only" tree.
phylo-only solution:
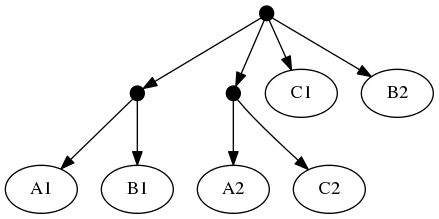
That solution displays both of the groups from the input trees, but lacks any of the 3 higher level taxa.
Note that the tips of the input trees need not be mapped to a terminal taxon in the pruned OTT.
The interpretation of such tips is not always clear. We assume the following of a tree, t, that has a tip mapped to a higher taxon Y:
- The presence of a
Yas a tip intshould not be taken as evidence of the monophyly of the taxon. - If building the supertree with all of the inputs except for the fact that
Yis pruned fromtreturns a supertree in whichYis monophyletic, then we say that the analysis supports the monophyly ofY. Note that this is often easy to check by performing small scale synthesis only trees that support or reject the monophyly ofY. - If the analysis without
Yintsupports the monophyly ofY, then replacingYwith a polytomy of all of the contained tips underYconveys the same phylogenetic information astwith the exception of the fact that the "taxonomically expansion" appears to provide support for monophyly. - If the analysis without
Yintrejects the monophyly ofY, then the input tree withYas a tip label is difficult to interpret. We assume in these cases, it is acceptable to pruneYofftbecause that information fromtis ambiguous.
We will consider adopting a more aggressive form of pruning to avoid ambiguous inputs: if any of the phyloenetic trees reject the monophyly of the taxon Y, then we will prune Y from t.
This decision is convenient, but also justifiable because the algorithm has detected that there is some controversy about the meaning of the input.
If no tree rejects the monophyly of the taxon, then the taxonomy will support its monophyly. So the lack of reject of monophyly by a phylogenetic input implies support for monophyly from the full analysis (which includes the taxonomic tree).
Conjecture 1: The set of equally optimal trees (A) can be expressed as a forest of rooted phylogenetic networks, but (B) may not necessarily be expressable as a tree.
We will use the phrase "phylogenetic graph" to refer to a direct graph in which each connected component in which each connected component is a phylogenetic network (contains no directed cycles).
One can think of an incompletely resolved tree (a tree with polytomies) as representing the set of trees that can be obtained by fully resolving the polytomies (but retaining all groupings in the original trees).
The proof of (B) is straightforward. Consider the inputs:
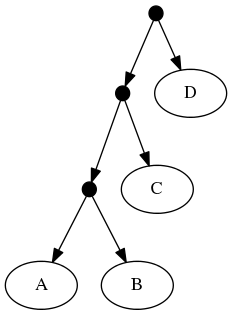
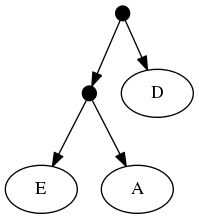
The follwing set of trees that can be produced by taking the union of all resolutions of both of these trees:
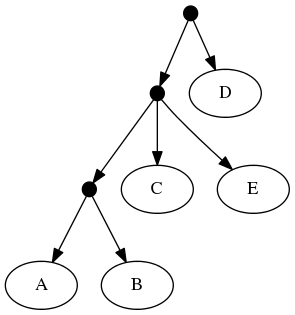
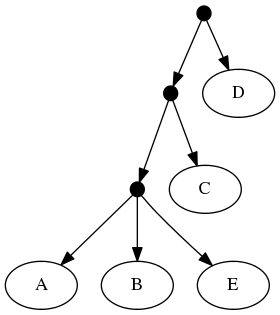
Or by the phylogenetic network:
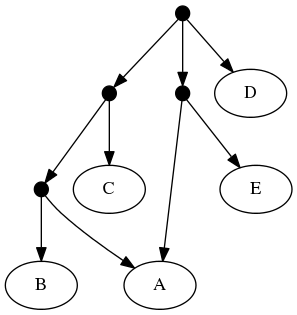
But not by the set of all trees that resolve the:
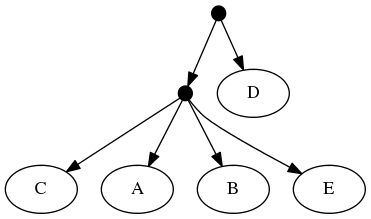
or any other tree because no tree displaying the rooted triplet ((A,C),B) is a valid solution.
We hope to prove part (A) of the conjecture by describing an algorithm.
Consider the case of an clade or single tip referred to as X is present in an input tree, t, but:
- no taxa assigned to
Xa have been added to a directed phylogengetic network,N, and -
Nhas all of the other taxa from inta connected component. - the edge that connects
Xto the rest oftis not a edge adjacent to a root with out-degree=2.
The procedure that we will call "phyloreference placement" of X consists of:
- generating a phylogenetic definition of the node or edge that
Xattaches to intwhich uses as designators all of the relevant leaf taxatexcept the taxa underX. This could be the definition of an edge (if the parent ofXis bifurcating, or a node if the parent ofXis a polytomy). In the case of a node definition the phyloreference is a statement of all of the taxa that descend from the node. For an edge definition, the descendant taxa of the edge and the "outgroup" are all included in the definition. - looking for a corresponding edge, path, or node in
Nbased on the definition: 1. if an edge is found,Xis added to theNcreating a parent for it that bisects the edge; 2. if a node is found, then it is treated as the parent toX3. if a path is found, thenXis attached by creating a loop from the start node of the path to the end node and and adding a parent forXalong that path. 4. if the object defined by the phyloreference forXdoes not exist, then the descendant leaf set of the grandparent ofXis calculated. The LICA of these leaves inNwill serve as the parent of theXwhen it is attached to the graph.
If there exists a node X in one input trees, t such that all of the leaves correspond to taxa which are not found in any of the of the input trees other than t, then we call the group defined by X a "single-source clade".
If we were to find the phylogenetic graph that represents the optimal solution it would be identical to the solution that one could obtain by:
- pruning the single-source clade from the input tree,
- finding the optimal phylogenetic graph for the pruned inputs,
- using phyloreferencing placement to attach
Xto the graph.
Conjecture 3: Building the phylogenetic graph by adding one tree at a time produces the optimal solution.
Because of the strong preference for groupings from trees of high importance, the algorithm does not need to consider the downstream consequences of adding a split. A greedy approach works because the addition of weight to the score from satisfying one edge from a high importance tree is larger than the score benefit from adding all of the groupings from trees of lower importance.
note: should also demonstrate that the order that you try to add groups from within the same tree does not have side effects - in the sense that any order of additions of groups from a tree to a network will result in the same output. This seems intuitive because all of the groupings within the same tree are compatible. But I am not certain if it holds, given that we are adding to phylogenetic network (rather than a tree).
See this page for one discussion of a scoring criterion which MTH thinks could be viewed as a criterion that the automated "tree synthesis" attempts to solve. This page will discuss some algorithmic considerations about this style of synthesis.